
Anke Schumann, MD/PhD
Metabolic pediatrician
Medical head of the Laboratory of Clinical Biochemistry and Metabolism
European Certificate in Pediatric Metabolic Medicine
E: anke.schumann@uniklinik-freiburg.de
T: +49 761 270 43730
F: +49 761 270 45270
Medical Center– University of Freiburg
Center for Pediatrics
Department of General Pediatrics, Adolescent Medicine and Neonatology
Mathildenstr. 1
79106 Freiburg
Germany
Our research focusses on pathomechanisms driving kidney disease in different metabolic diseases (e.g., propionic aciduria, Fabry’s disease) using human model systems. Our special interest lies on the crosstalk between different organelles and cellular compartments. We also aim at the identification of potentially targetable aims to modify affected pathways and the evaluation of specific compounds on renal cellular energy metabolism.
TEAM
Impact of diet on renal phenotype in propionic aciduria
Véronique Belche (MD student)
Renal tubular involvement in Fabry’s disease
Kristin Schaller (MD student)
Modification of renal cell metabolism in health and disease by different compounds
Katharina Klotz (Technician in training)
Impact of diet on renal phenotype in methylmalonic aciduria
Marion Brutsche (MD student)
RESEARCH THEMES
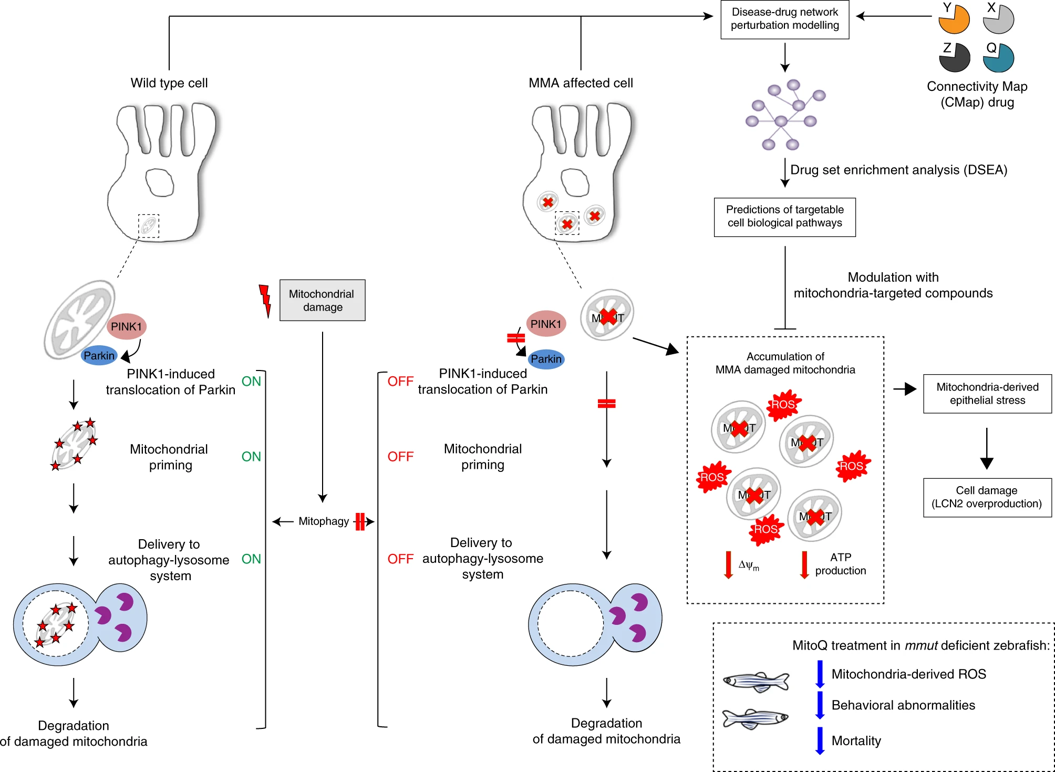 Inborn errors of energy metabolism (IEM) are monogenic defects typically presenting with acute, potentially live threatening metabolic crisis in the neonatal period. Due to improved medical care patients grow older and long-term complications emerge, selectively affecting only specific organs, while the defective enzyme is expressed in all tissues.
Inborn errors of energy metabolism (IEM) are monogenic defects typically presenting with acute, potentially live threatening metabolic crisis in the neonatal period. Due to improved medical care patients grow older and long-term complications emerge, selectively affecting only specific organs, while the defective enzyme is expressed in all tissues.
The kidney is a target organ in several IEM. Based on the hypothesis that its vulnerability is based on high energy demands, we aim to elucidate how different defects impact on renal cellular homeostasis.
Our group is currently working on projects addressing the pathophysiology of chronic kidney disease (CKD) in mitochondrial and lysosomal disorders using Propionic aciduria and Fabry disease as an example.
We established and characterized human renal tubular epithelial cell lines of patients and healthy controls as tissue-specific models. The aim of the investigations is to understand how the impairment of different organelles leads to a disruption of the cellular mitochondrial (energy) metabolism and which (intra-) cellular communication pathways are modulated in a disease-specific manner. Of note, both diseases have therapeutic interventions (e.g., protein reduced diet and enzyme replacement therapy (ERT)) both of which are not curative nor sufficiently hindering the progression of CKD. A better insight into underlying pathomechanisms might point towards new therapeutic options and help to identify potentially targetable aims with high translational potential.
Specifically, we aim to investigate:
- mitochondrial morphology and function
- mitochondrial quality control (mitophagy/autophagy)
- mitochondrial dynamics (fission/fusion/mitochondrial biogenesis)
- intracellular trafficking and transport
- the impact of pharmacological compounds/diet (e.g., effect of low protein diet/ERT, anti-oxidants, modulators of autophagy, modulators of mitochondrial energy metabolism on aims 1-4)
in human, tissue-specific models and will finally evaluate our findings by metabolomic and proteomic analysis of human urine and plasma samples on a systemic level.
SELECTED RECENT PUBLICATIONS
For a complete publication list for Anke Schumann, please click here: https://pubmed.ncbi.nlm.nih.gov/?term=Schumann+Anke
FUNDING
Research award of the German Society for Pediatric Metabolic Diseases
Nutricia metabolic research grant